

Biosimilar medicine is a biologic product. The US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) define a biosimilar product with certain differences. According to the FDA, “a biosimilar is a biologic medication that is highly similar to and has no clinically meaningful differences from an existing FDA-approved biologic, called a reference product. Compared with a reference product, biosimilars are made with same types of living sources, are given to the patient in the same way, have the same strength, dosage, potential treatment benefits and potential side effects.” As per the EMA, “a biosimilar is a biological medicine highly similar to another biological medicine already approved in the EU in terms of structure, biological activity and efficacy, safety and immunogenicity profile.
The difference lies in the requirement of data submitted to the FDA and the EMA. In the US, data demonstrating the biosimilarity includes, analytical studies, animal studies, and data proving safety, purity, and potency. In the EU, an applicant must demonstrate the biosimilarity through comprehensive comparability studies and it varies on case-by-case basis.
Biosimilars have undeniable benefits that they offer across different stakeholders and drive healthcare towards a more accessible, equitable, and sustainable future. They are highly important for a number of reasons, such as:
a) Research and development cost of biosimilars is less than the innovator drug.
b) Enable increased market competition and tackle monopoly.
c) Cost-saving treatment methods to the patients.
d) Reduced financial burdens on the overall healthcare system.
e) Significant price reductions offered by biosimilars allow healthcare systems to distribute budget resources more efficiently, potentially increasing the overall coverage for new and innovative treatments, better public health outcomes and population well-being.
Biologic market and biosimilars are gaining traction due to the looming patent cliff in the coming decade which will affect many blockbusters. The biosimilar market in Europe reached US$ 11,849.5 Million in 2023. Looking forward, it is expected that the market reaches US$ 53,222.9 Million by 2032, exhibiting a growth rate (CAGR) of 17.6% during 2024-2032. The US biosimilars market size was valued at $8.6 Bn in 2022 and is estimated to reach $35.1 Bn in 2030 with a CAGR of 20.2% from 2022 to 2030.
The Biosimilar approval pathway came into place in the EU in 2003 whereas the biosimilar pathway set out in the Biologics Price Competition and Innovation Act (BPCIA) of the US became active in 2010, marking a total of 7-year gap from the EU. In line to this, the EU market witnessed first biosimilar approval in 2006 in the form of Omnitrope whereas the first FDA approval was seen in 2015. Till date, there are 80 biosimilar products approved in the EU and 46 biosimilar (interchangeable inclusive) products in the US (Figure 1). The majority of biosimilar approvals in the EU include the drug classes of immunomodulators and anti-neoplastic agents (Figure 2).
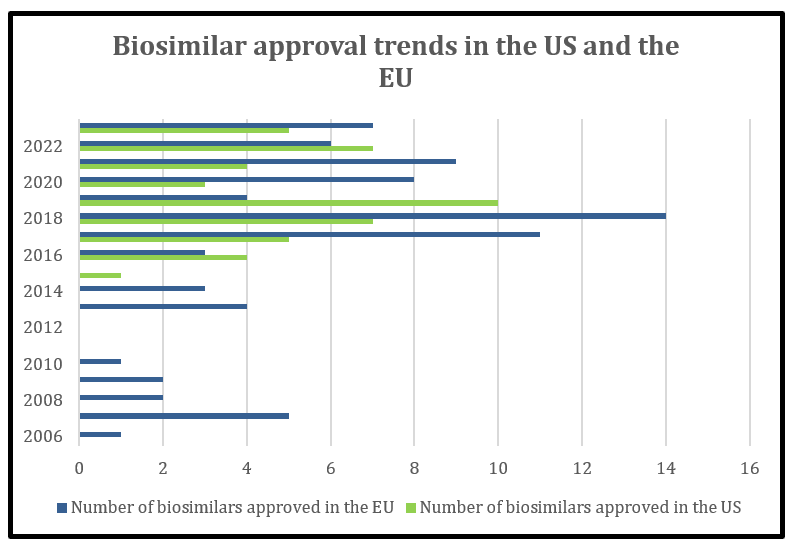
Figure 1: Comparison of number of biosimilars approved in the US vs EU
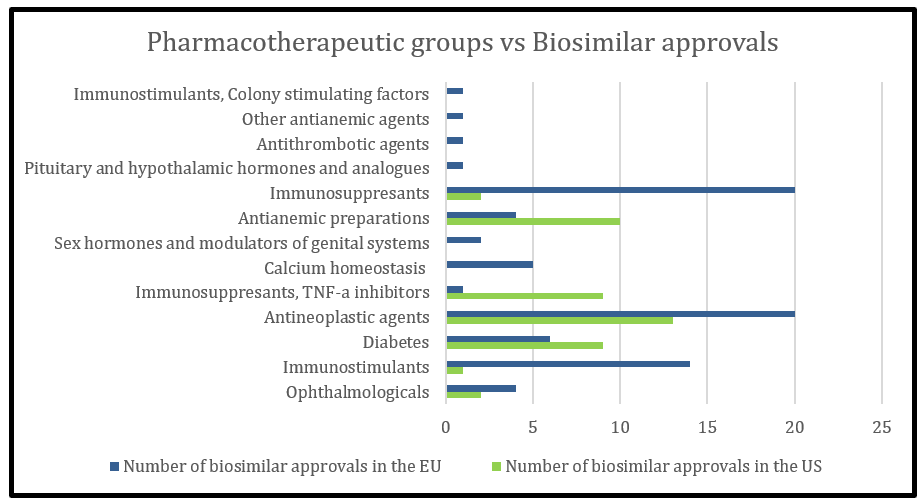
Figure 2: Pharmacotherapeutic groups of biosimilar approvals approved in the US and EU
In the US, the BPCIA also defines an interchangeable biosimilar as a biosimilar that is expected to produce the same clinical result as the reference product in any given patient. It may be substituted for the reference product without prescriber intervention, and the risk in terms of safety or efficacy of switching or alternating between biological products is no higher than using the reference product alone. Thus, “interchangeable” works as an added designation to a biosimilar product. There is no such requirement or designation in the EU.
In the US, the biologics data exclusivity is 12 years from the date of first approval and is a combination of Biologics Application Submission Exclusivity and Biologics Application Approval Exclusivity (BASE and BAAE) whereas in the EU, the protection period is of 8+2 years. However, the European Commission has proposed adjusting this to a basic 6+2 regime, by reducing the period of data exclusivity by two years, with a possibility to extend this by up to four additional years, if certain criteria are met. These criteria are: when the product addresses a “high unmet medical need,” an additional six months will be available; for additional therapeutic indications with significant clinical benefit there will be a further 12 months’ exclusivity; or if, for new active substances, comparative clinical trials are conducted, this will result in an additional six-month period of exclusivity. Finally, in order to encourage the availability of pharmaceuticals in all member states, by sufficiently marketing the product in every authorized EU state, a further 24 months of exclusivity can be obtained. When this proposed law substantiates, it may further enhance the biosimilar products approval in the EU.
With respect to the patent landscape in the two continents, it has time and again been observed that the EU patents expire earlier while the innovator companies may try to generate more patent barriers in the US. The prolonged litigation suits that finally end up with settlement between the parties may be one of the reasons that delays the biosimilar market entry into the US.
All these factors contribute to significant lesser biosimilar approvals in the US as compared to the EU. But these are not limiting. There are a few other reasons that may further cause reluctance in the commercialisation of biosimilar products. Some of these may include:
- Price point: In the US, there is a huge fragmentation in the payer healthcare system and their perception of safety and efficacy. This is driven by pricing and rebates. The interplay between manufacturers, insurers, and Pharmacy Benefit Managers (PBMs) play a huge role in stalling price reductions. In the EU, on contrast, there is a single-payer healthcare system and they prioritise cost- effectiveness. The biologics, in EU, are often procured through tenders and biosimilars thus win by offering low prices.
- Perception: The US physicians are still uncomfortable in prescribing biosimilars. This might be due to the fact of unawareness related to the safety and efficacy similarity between the two products. Also, patients are sceptical in adopting biosimilar treatments due to similar unawareness and price comparisons.
Nonetheless, it seems that the US is on the verge of a major shift in this paradigm. To increase biosimilar approvals, market penetration and uptake by prescribers, the FDA released a biosimilar action plan (BAP) in 2018 which focusses on following key areas:
a) improving the efficiency of the biosimilar and interchangeable product development and approval process.
b) maximizing scientific and regulatory clarity for the biosimilar product development community.
c) developing effective communications to improve understanding of biosimilars among patients, providers, and payers.
d) supporting market competition by reducing gaming of FDA requirements or other attempts to unfairly delay market competition to follow-on products.
Another ground-breaking method is the introduction of Inflation reduction act (IRA) of 2022. The Bidden administration announced the first 10 drugs to have their prices negotiated in 2026 by Medicare on 29th August 2023. Of these 10 drugs, 3 are biologics: Enbrel, Stelara, and Fiasp & Novolog. The manufacturers have also agreed to these negotiations. This may further impact the cost reduction of follow-on products. These were selected because of the absence of their biosimilar versions. However, Wezlana, an Ustekinumab biosimilar, was approved in October 2023. It would be interesting to see how Amgen commercialises Wezlana and how it impacts the the ‘maximum fair price’ negotiation. It may also be possible that Stelara be deselected because of the biosimilar approval. This is just an example of how IRA might play an important role in the shuttle between increasing biosimilar approval and manufacturers hesitancy in price reductions.
Some other methods to facilitate faster market entry can be taken into consideration, such as, policy makers may consider limiting the patent prosecution, compulsory patent listing in the “Purple book”, and enhanced antitrust enforcement.
References:
- https://www.imarcgroup.com/europe-biosimilar-market
- https://bionest.com/biologics-us-vs-europe/
- https://www.iconplc.com/insights/blog/2020/05/27/the-differences-between-the-us-and-eu-biosimilar-markets
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7824407/
- https://www.hcplive.com/view/breaking-down-barriers-why-biosimilars-face-resistance-us-market
- https://www.bigmoleculewatch.com/2023/11/06/fda-approves-amgens-wezlana-as-biosimilar-to-and-interchangeable-with-janssens-stelara-ustekinumab/